���q�O���@���s�����߂�Linux�̓����@�i�����܂ŎQ�l�ł��G�@�w���̍��͎���܂�قǃv���O���~���O���M�V�M�V����Ă��܂������A���͂̂�т�PC������Ă��܂��j
-�@�P�̖ڕW�́@Linux�ŕ��q�O���@�����@-�@�i�ŏI�I�ɂ͒EMS-Windows�j
�@�@���╪�q�O���@���茳�ɂ���o�b�Ōv�Z�ł���I�@40�N�O��w�̎��A��^�R���s���[�^�Ő��x�̈������q�O���@
Extended �g������e�� method �Ōv�Z���Ă����̂Ɣ�ׂĂ����Ԑi�������̂ɋ����Ă��܂��B�@�t���������ł�낤�Ƃ��Ă���
�b�m�c�n/�l�h�m�c�n �ł̍��̕��q�̕��q�O���v�Z���ł���悤�ɂȂ��Ă���I�@���@�v�Z���邽�߂�
�k�h�m�t�w ���g���Ă݂悤�Ƃ������Ƃ���n�߂܂����B�@�f��������������
�ło�b�̃f�B�X�v���[�ɕ��q��`���ĉ�]�����Ē��߂�E�E�E����Ȃ��Ƃ���v�������Ƃ�����܂��B�@�܂��ALINUX
�͂قƂ�ljp��̐��E�Ȃ̂ŁA���̐��E�ɓ����ĉ��Ƃ��m���Ă�낤�Ǝv���Ă���Ă�����p��͂��������邩������܂���B�V�y�̏ꍇ�A�V���h�~�ɂȂ邩������܂���B
�@�@���������āA���̃y�[�W���J�����l�ɉ��w��Ƃʼn��w�������̊J��������Ă���l�������ꍇ�A���R�@�Ő�����������\������̂ɂl�n�o�`�b�v�Z���g���Ȃ����@�Ƃ������ꂪ����܂��i2000�N��͘b������オ�����悤�ł��j�B�l�n�o�`�b
�̎g������m���đ��͂���܂���B�������������g���[�X����ɂ͎��Ԃƃe�N�j�b�N���K�v�A�������@�ɂ��ĕ�����ǂ�ł��ԈႢ���w�E�ł��邩�����Ȃ��Ƃ�����܂��B�����̓_���J�o�[�ł���̂��l�n�o�`�b���̕��q�O���@�ɂ��v�Z�ƍl�����Ă���悤�ł��B�������h���b�O�f�U�C���̈���@�Ƃ��Ă��g���܂��B
�@�@�y��̏Đ����l�n�o�`�b�Ōv�Z����ڍׂȕ��@�ɂ��Ă͕ʕłɏ����܂����B�����Ȑl���̌��ł���悤�AMS�|Windows�łł���悤�A�O���t�B�b�N�c�[����WINMOSTAR��p���AMOPAC7��WINMOSTAR���玩���I�ɓ��������@���Љ�܂����B
�P�D�V�X�e���̍\�z
�@1-�P�D�@Linux��p��PC����
�@�@�@�{�̂T���~���x�̈����ȃf�X�N�g�b�v�o�b���g���B���R�͉��L�B
�@�@�@�@�@MS-Windows��virus��PC�ɔ�Q���y�ڂ����Ƃ��x������A���̋t���^
�@�@�@�@�@MS-Windows�̃R�}���h�v�����v�g�œ������Ƃ����悤�Ȉ��Ղȕ����ɂɍs���Ȃ��i�ǂ����Ă�MS-Windows�ł�肽���l�ɂ͍����Ă��邪�j
�@�@�@�@�@�V�X�e�������Ȃ������悤�ɂȂ������LINUX��MS-Windows�̃f���A���u�[�g�ɂ��Ă�������
�@�@�@�@�@MS-Windows�̃R�}���h�v�����v�g�œ������ƊȒP�Ȃ悤�ň��
path �ɖ�肠��iWinmostar���g���n�߂�����Ȃ������܂����BWinmostar��
install �ׂ͍����Ƃ���܂ł���Ă��ꂽ�悤�ł��B�j
�@�@�@�@�@�@���@��p�o�b�Ƃ��ăh�X�p����Regulus�iCPU�FAMD�j���w���A�f�B�X�v���[�͉ƒ�pTV�i�P�g���C�p�j���g�p�i�摜�̎��͂�∫���APC���g�����͂s�u������Ȃ��Ƃ������_�͂���܂����j
�@�@�@�@�@�@�@�@�i20�N�O�͎���o�b�Ƃ��Ĉ����茾�t�ɔ̔����Ă��܂������ŋ߂͐M�������[���ɂȂ��Ă���悤�ł��B�P���ɂT���Ԉȏ�@�Ђǂ����͂Q0���ԋ߂��@�Q�N�ȏ�g���Ă��܂����n�[�h�I�ȃg���u���Ȃ��B�j
�@�@�@�@�@�@���@�����A�r�f�I�J�[�h�����Ȃ��Ŏg���Ă��܂������iCPU�ɓ�������Ă���O���t�B�b�Ndevice�ł���Ă����j�ALinux�ł���t�����������͓����܂���ł����iD�����������͓����܂����j�B��肪�������悤�ł��B�����ȁi3000�~��̔ėp�j�r�f�I�J�[�h����ꂽ��UBUNTU�������Ȃ�悤�ɂȂ�܂����B
�@1-2..�@�f�B�X�g���r���[�V�����̑I��
�@�@�@�n�r�ł���Linux���ȒP�ɓ��������߂̃\�t�g���f�B�X�g���r���[�V�����ƌ����Ă���
�@�@�@���̂��Ƃ�OS�ł���l�r-�c�n�r�ło�b�����Ă���\�t�g�ł���MS-Windows�Ɠ���
�@�@�@LINUX�ɂ͐��\�̃f�B�X�g���r���[�V����������
�@�@�@���y�̓f�B�X�g���r���[�V�����Ƃ��āu�c�d�a�h�`�m�v��I�A���̗��R��
�@�@�@�@�@�p�P�[�W�i�ȒP�ɃC���X�g�[���ł���\�t�g�j���L�x�A�ݒ�Ɋ��̋K�肪�ɂ₩
�@�@�@�@�@Puppy�̂悤�Ȍy������ޭ���݂����ذ�ޓ��������M�������Ⴍ�AUbuntu�̂悤�ȏd���f�B�X�g���r��-�V�������Ɛݒ�ɋK�肪�����iUEFI/ڶ�̖��j
�@�@�@�ި����ޭ���݂�UBUNTU������܂��BUBUNTU��ݽİق��Ďg���Ă݂܂������A���q�O���@�iMOPAC�j�̓��o�͂��s�����߂̃O���t�B�b�N�c�[�������܂������Ȃ��A�߯���ނɕ��q�O���@�֘A�̃\�t�g�����Ȃ��@�Ƃ�����肪����A�C���X�g�[����P�T�ԂŃA���C���X�g�[�����܂����B���t����������
�� �c���������� �� �������������� / uninstall
������Ă��܂����t�����������ł͎v���Ă���悤�ɂ͓����܂���ł����B���q�O���@���g��Ȃ���t�����������͂����̂ł����A���y�̂悤�ɕ��q�O���@���C�����Ƃ����Ă��܂���B�t�����������Ŗ��Ȃ������O���t�B�b�N�c�[����T�������̂�������܂��B�i�k���������̕\�v�Z/ܰ���/�ް��ް�/���{��ϊ�
�\�t�g��MS-Windows�̿�Ă��g�����ꂷ���Ă��鏬�y�ɂƂ��ċ@�\�����Ȃ����Ă��܂��B�֑��ł����AMS-Windows�̓��{��ϊ��\�t�gIME�͋��`�h��ĎЂ̂`-WAORD��WX��Ă��������̂Ƃ����\�������Ƃ�����܂��B������������̺��߸Ăȿ�Ăł����BMS�Ђ͂R�j�Ђ́u���v��Just
Systems�Ђ́u�ꑾ�Y�v�̋Z�p�ł͂Ȃ�AI�Ђ�WX��]�������Ƒz�����Ă��܂��B�j
�@�@�@2017/4���_�ŁA�k�������� PC��D�����������Ƃt�����������̃f���A���u�[�g�ɂ��Ă��āA���X�t���������������Ă��܂��BUbuntu���g���Ă���ƁA�o�b�N�O���E���h�ŏ��������Ă��邱�Ƃ������ē��삪�ɒ[�ɒx���Ȃ邱�Ƃ�����܂��B�c�����������ł͂��̂悤�Ȃ��Ƃ��Ȃ��B���q�O���@�̂悤�Ȍv�Z�����ɂ͂c�����������������Ă���Ǝv���Ă��܂��B�c���������������������͎��Ӌ@��Ƃ̐ڑ��������_�ƗV�ѿ�Ă���r�I���Ȃ��_�B�������d��������t������������������������܂���B���y�A����/������͂c�����������Őݒ肵�Ă��܂���B�c�����������Ƃt�����������Ńf�[�^�����L���ł���̂Ŗ��Ȃ��Ǝv���Ă��܂��i���߰հ�ް�̌������K�v�ł����j�B
�@�@�@Debian��version�ɂ���Ďg�������\�t�g���߯���ނɂȂ��Ƃ��������Ƃ������āA���y�����܂��g����version���ǂ������Ƃ������Ƃ�����܂����B���y���g���Ă���̂�version7.8.0
�@�@�@�߯���ނƂ��āusynaptic�v���C���X�g�[�����Ďg���t�a�t�m�s�t�ł��قȂ�version��Debian�ł�ݽİقł��邱�Ƃ��킩��܂������A�t�a�t�m�s�t�ł̓C���X�g�[�������\�t�g�������Ȃ��Ƃ������ʂł����B
�@1-3..�@�Z�L�����e�B
�@�@�@�C���X�g�[�����ɈÍ������邱�Ƃ����߂܂��B�I���ňÍ������C���X�g�[���ł���悤�ɂȂ��Ă��܂��B
�@�@�@�c���������������߂�t�@�C�A�E�H�[���̐ݒ���ꉞ�K�v�ł��BLinux�͊ȒP�ɂ͐N���ł��Ȃ��悤�ɂȂ��Ă��܂����A���y�̓\�t�g�����������g���Ă��܂��B
�@�@�@���i�N�b�X�ɂ͑����̃Z�L�����e�B���@������܂��i�Ʊ�������ق܂ł���܂��j�B�[���̂������x���܂ł���Ă��������B
�@�@�@���y�͕��q�O���@�ɊW����\�t�g�������i�b�N�X�̂o�b�Ƀ_�E�����[�h����悤�ɂ��Ă��܂��B�@�@�@�g���n�߂�2�N�������q�O���@�ň�i��A�����̂ł������̃\�t�g�������Ă��܂��B
�@�@�@���[�������i�b�N�X�o�b�ŊJ���邱�Ƃ�����܂����A�����Ƃ��ēǂނ����B���M����Ƃ��͏������e�L�X�g�Ƃ��đ����Ă��܂��B
�@1-�S..�@�O���t�B�b�N�c�[����Ghemical��I��
�@�@�@MOPAC���̗ʎq���w�v�Z�\�t�g�͌v�Z�����ō��W���́A�v�Z���ʂɃO���t�B�b�N�\�����ł��Ȃ�
�@�@�@Ghemical��MOPAC���̓���/�o��°قƂ��Ďg����
�@�@�@���o���I���q�O���@�����łȂ��Aab initio�A�ʎq�͊w�̓��o�͂��ł���
�@�@�@�ݒ肪�e��
�@�@�@�R�}���h�������I�Ń}�j���A��������قǓǂ܂Ȃ��Ă���������B
�@�@�@�A���A�o�O�������āA���ꂪ���N�C������Ă��Ȃ��悤�Ō��E�̉\������A���E�ɑ��������ꍇ�A���̃c�[����T��
�@�@�@�i����̃\�t�g�ɑΉ��ł���悤��X�o�̓t�H�[�}�b�g���p�ӂ���Ă��邪�A�t�H�[�}�b�g�I���Ɏ�Ԃ��������Ă���
�� ���y�̏ꍇMOPAC�̃t�H�[�}�b�g�������K�v�Ȃ̂ŁA�@�\�����������邱�Ƃ��������F�\�t�g���������y���݂�Linux�ɂ���܂��G���y�A��w�����Fortran�Ńv���O���~���O�����邱�Ƃ��V�тł����B��ƂɏA�E��A��X�\�t�g�̃}�N��/�u�a�r/�r�p�k/Basic����邱�Ƃ����ʂ̐l���Q�[�������̂Ɠ����V�тł��܂������j
�@�@�@2017/4�̎��_�ł͂f�������������Ɍ��E�������Ă��܂��A�t���[�E�F�AAbogadro���C���X�g�[�����Ď��X�g���Ă��܂��B�g������͂܂��܂��B�����_������Ώ�芷���邩������܂���B�܂��A�r���A�[�Ɍ��h���������̂�����܂����A�v�Z���ʂ̐��l�ɊS������̂Ŏg�����Ƃ͎v��Ȃ��i�K�ł��B
�@1-�T.�@���q�O���@�Ƃ��Ăl�n�o�`�b�V��I��
�@�@�@�������x���ł͈�̑O�̔��o���I���q�O���@�̂`�l�P/�o�l�R�ŏ[��
�@�@�@free ware��M�nPAC7��AM1/MIND0/PM3���ł���@�iMOPAC�ł�MINDO��MNDO��"�h"���Ȃ��`�Ŏg���Ă��܂��j
�@�@�@�������i�݁A�l�n�o�`�b7���s�[���ƍl����ꂽ�ꍇ�A�L��MOPAC��ab
initio�A����PC��p�������
�@1-�U.�@���q�O���@�����ɖ𗧂Ă�H
�@�@�@�i�������炪�l/�w�Z/��Ƃ̑n����/���f�j
�@�@�@�y��Â���ŕ��q�O���@���g�����Ƃ��l����ƁA�Đ����x�ƏĐ����Ԃ̊W�B�S�y�̑�\�I�����E���̒E���������v�Z����H�@����ȕ��ɕ��q�O���@���l���Ă��܂��B
�@�@�@���y�̋����͌v�Z�̌��ʂł͂Ȃ��A�v���O���~���O�B�O���t�B�b�N�\�t�g�╪�q�O���@�\�t�g�����ǂ��邱�ƂȂ̂ŁA������Ɍ������Ǝv���܂��B���ʂ̐l�͌��ʂɋ���������Ǝv���̂ŁA�ʔ������ʂ��o�����Ƃ����܂��B������S�������Ȃ����Ƃ�����Ă��d�����Ȃ��̂ŁB
2.�@�X�\�t�g�̃C���X�g-��
�@2-1�@Debian��install
�@�@�@MS-Windows�œ����Ă���o�b��Debian
install�p�b�c���Z�b�g�����
�@�@�@CD��install�pDebian�����t����
�@�@�@PC��boot��CD���グ�ɂ���PC�𗧂��グ��@�iDebian�̏ꍇ�A���K�V�[�ʼn\�j
�@�@�@�@�g�c�̂���Debian �i�k�h�m�t�w�j�̈��ݒ肷��i�S���Linux�ɂ��邱�Ƃ����߂�G��ʓIDebian�����ɂ͕ʂ̿�Ăŗ̈�m�ۂ����߂Ă���j
�@�@�@�@install���I��������A�c�����������̏��@�\���g����悤�ɂȂ��Ă��邱�Ƃ��m�F
�@2-2�@Ghemical��install
�@�@�@Debian���߯���ނ�Ghemical�����邱�Ƃ��m�F
�@�@�@Ghemical���߯���ނ�install����i�֘A��Ă�������install�����j
�@2-3�@M0PAC7��install
�@�@�@Debian�����߯���ނ�M�nPAC7�����邱�Ƃ��m�F
�@�@�@Mopac7���߯���ނ�install����i�֘A��Ă�������install�����j
3.�@�f�������������̎g����
�@3.1.�@Ghemical�̓��{��}�j���A���@
�@�@�@Ghemical��հ�ް�ƭ�ق̓��{���������܂��@�i����I�Ȃ̂��ȒP�ƭ�ق͂������N���b�N�j
�@�@�@�i�����܂ŎQ�l�ł��B�v�n�q�c���ɓ\��t���āA�摜�����{����R�s�[���āA�ԈႦ�Ȃǂ��C�����Ďg���Ă��������B�j
�@�@�@�i���{��Ghemical��"�߯����"�ɓ����Ă��܂��B�j
�@�@�@�i���y��PC�̏ꍇ�A̧�ق̼���/usr/share/ghemical/3.0.0/user-docs
html��install����Ă��܂����B�A���Aver.2.00���ƭ�قł����j
4.�@�l�n�o�`�b�V�̎g����
�@4-1�@�l�n�o�`�b�V�̓��͕��@
�@�@4-1-1. �L�[���[�h�̓���:Z-matrix���͗��̏�R�s�̑�P�s��
�@�@�@�@�@1) M0PAC7�̋@�\�̂ǂ�Ōv�Z���邩���w�肷��B�Ⴆ�η�ܰ�ށuPM3�v��PM3������Ʊ݂��g���B
�@�@�@�@�@2) �����̷�ܰ�ނ͋ŋ��
�@�@�@�@�@3) �啶��/�������͂ǂ���ł��@�\�͓���
�@�@�@�@�@4) �P�s�Ŏ��܂�Ȃ����́A�ŏI���������\���Ƃ��邱�ƂŎ��s����ܰ�ލs�ɂȂ�
�@�@�@�@�@5) ��ܰ�ނ̎g�p���@�̓}�j���A��/�g�p����Q�l�ɂ��Ď��s���낷��
�@�@�@�@�@6) �悭�g�p����鷰ܰ��
�@�@�@�@�@�@�@�@PM3
����݂Ƃ���PM3�Ǝw��iMOPAC7�ł�AM1�AMINDO/3�APM3������݂Ƃ��Ďg����j
�@�@�@�@�@�@�@�@PRECISE
SCF�I����臒l�����������Čv�Z����
�@�@�@�@�@�@�@�@SYMMETRY ���q�ԋ���/�����p/��ʊp�ɂ��đΏۂɌv�Z�i����6�ɓƗ����Ƃ��ċL�q�j
�@�@�@�@�@�@�@�@FORCE
�U���v�Z���s���J�ڏ�Ԃ���������B���ʂ�xyz�����̐U�����̒l�Ƃ��ďo��
�@�@�@�@�@�@�@�@TS
���Ѵ�ٷ��߽�ŊT�Z���A�T�Z�l�������l�Ƃ��Ă��̷�ܰ�ނőJ�ڏ�Ԃ����߂�
�@�@�@�@�@�@�@�@SADDLE
�J�ڏ�Ԃ�T�����邽�߂̏����\����T���BSADDLE�v�Z�ɗp����2��Z-matrix�͓�������/���������l���łȂ��Ă͂Ȃ�Ȃ��B�uSADDLE�v�Ɓuxyz�v�p����̂���ʓI
�@�@�@�@�@�@�@�@VECTORS�@�@�@�ŏI�ŗL�x�N�g����\������@�i����ŗL�G�l���M�ɂ�����d�q���x�̕��z�̕\��
�G �n�[�g���[�}�g���b�N�X�s��̉��ł���x�N�g���j
�@�@�@�@�@�@�@�@BONDS�@�@�@�@�@�ŏI�����x�}�g���b�N�X��\������@�i�e���q�Ԃ̓d�q���x��\���}�g���b�N�X�j
�@�@4-1-2. �R�����g�̓���:Z-matrix���͗��̏�R�s�̑�2�A3�s��
�@�@�@�@�@1) M0PAC7�̋@�\�ɉe�����邱�Ƃ͂Ȃ�
�@�@�@�@�@2) �v�Z���e��\���L�q������
�@�@4-1-3.�@���q�\�����W���̓��́F�@
�@�@�@�@�������W�œ��́@�i��ü�ݍ��W�ixyz�j�œ��͂��邱�Ƃ��ł��邪�A���̕����������ē���j
�@�@�@�@���f��/���W/�ׯ��/�����W�������z���
Z-matrix �ƌĂ�ł���B
�@�@�@�@���Ɏ����̂�MOPAC�l���� Z-matrix�B�@�Ⴆ�AGAMESS�ɂ�GAMESS�l����
Z-matrix ������B
�@�@�@.�@�@1�j�@ Z-matrix��i��͔���₷���悤�ɑ����Ă��邪�A�����ް��͋ŋ���Ă���iTab�ł͂��߁j�����ŋ@�\����j�@
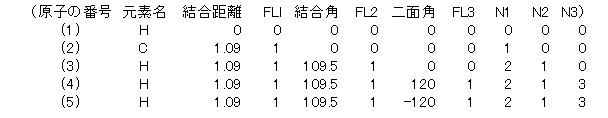
�@�@�@�@�@2) ���q�̔ԍ��ƌ�������/�����p/��ʊp�̓���
�@�@�@�@�@�@�@�@ �����̒��S�Ȃǂ̌��q���P�Ԃɂ���
�@�@�@�@�@�@�@�A �������Ă��鍜�i�̈���ɂȂ錴�q���Q�Ԃɂ��A���q�Ԃ̋��������œ��͂���
�@�@�@�@�@�@�@�B ���q�̔ԍ��Q�ƌ������Ă��錴�q�̔ԍ����R�Ƃ��A�Q�Ƃ̋����A3-2-1�̌����p����͂���
�@�@�@�@�@�@�@�C �S�Ԉȍ~�͓��Y���q-N1�̋����A���Y���q-N1-N2�̌����p�A���Y���q-N1-N2-N3������ʊp����͂���
�@�@�@�@�@3) ���f��
�@�@�@�@�@�@�@�@ M0PAC7���e����Ʊ݂����Ұ���p�ӂ��Ă��錳�f�̖��̂���͂���B
�@�@�@�@�@�@�@�A �v�Z��A���q�̍��W�ɖ�肪����ꍇ�A�_�~�[���q�i�w�j����͂��鎞������B
�@�@�@�@�@4) FL :�@�œK���ׯ�ށA���L�̐��l�̂ǂꂩ����͂���.�B
�@�@�@�@�@�@�@�@�@�@�\���œK���̎��͕��q�̑Ώ̐����l��������Łh1�h�𑽗p���邪�A�J�ڏ�ԂŌv�Z���鎞�͍\����ς��Ȃ����C�G�e�B�͍œK���ׯ�ނ��h0�h�ɂ���ȂǁA�œK���t���b�O���ǂ����邩�n������B
�@�@�@�@�@�@�@�@ ��1�@:�@�œK������
�@�@�@�@�@�@�@�A ��O�@:�@�œK�����Ȃ�
�@�@�@�@�@�@�@�B ���|1:�@���Ѵ�ٷް�߽�̌v�Z���s�Ȃ�
�@�@�@�@�@5) N1�AN2�AN3 : ���Y���q�ԍ��ȑO�̔ԍ����Q�Ƃ���
�@�@�@�@�@�@�@�@ N1 : ���������̑ΏۂɂȂ������q�̔ԍ�
�@�@�@�@�@�@�@�A N2 : �����p�̑ΏۂɂȂ������q�̔ԍ�
�@�@�@�@�@�@�@�B N3 : ��ʊp�̑ΏۂɂȂ������q�̔ԍ�
�@�@4-2. ���Ѵ�ٷް�߽�v�Z�̋����ϐ��l�̓��͓�:Z-matrix���͗��̉��ɍs����͂���
�@�@�@�@�@1) �J�ڏ�Ԃ�T������ۂɋ����ׯ�ނ�-1�ɂ��ĕ����̌��q�ԋ�������͂���
�@�@�@�@�@2�j�@�@�@�V�@�@�@�@�@�@�@�@�@�@�@��ʊp�ׯ�ނ�-1�ɂ��ĕ����̊p�x����͂���
�@�@4-3�@��ܰ�ށ@�uSYMMETRY�v�@�̎g����
�@�@�@4-3-1.�@����
�@�@�@�@�@SYMMETRY�f�[�^��Z-matrics�̌�ɒu�����B�����~�j�}���G�l���M�̔������W��SADDLE�̑�Q�f�[�^�̂悤��
�@�@�@�@�@���̃f�[�^������ꍇ�ASYMMETRY�̃f�[�^�͂����̌�ɒu���B
�@�@�@�@�@SYMMETRY�̃f�[�^�̕��ו��͎��̂悤�ł���
�@�@�@�@�@�@�@�@�w�茴�q�A�@SYMMETRY�W�A�@�w�茴�q�A�@�w�茴�q�A�@�w�茴�q�A�@�w�茴�q�A�@�E�d
SYMMETRY�W�@�@�@�@SYMMETRY�@�\

�@�@�@�@�@��L�W�̂���1�A2�A3�A14���ǂ��g����
�@�@�@�@�@SYMMETRY���g�����̓J�[�e�V�A�����W���g��Ȃ�
�@�@�@�@�@�W18�̓|���}�[�Ŏg�����߂ɍ��ꂽ����
�@�@�@4-3-2.�@SYMMETRY�̗�
�@�@�@�@�@�G�^�����Ƃ���SYMMETRY���L�[���[�h�ɂɂ����ꍇ�̓��͕��@������
�@�@�@�@�@SYMMETRY
�@�@�@�@�@Ethan�AD3D

�@�@�@�@�@3�@1�@4�@5�@6�@7�@8
�@�@�@�@�@3�@2�@4�@5�@6�@7�@8
�@�@�@�@����F �@���q�̔ԍ��R�Ɠ��������������q�̔ԍ�4,5,6,7,8�A�̌��q�ɐݒ肷��
�@�@�@�@�@�@�@�@�A���q�̔ԍ��R�Ɠ������p�����q�̔ԍ�4,5,6,7,8�A�̌��q�ɐݒ肷��
�@�@�@�@�@�@�@�@�B���q�̔ԍ�4,5,6,7,8�̌��q�̌�������/�����p/��ʊp�͕ς��Ȃ�
�@�@4-4�@��ܰ�ށ@�uSADDLE�v�@�̎g����
�@�@�@�@PM3/AM1/MNDO�ɂ���SADDLE�őJ�ڏ�Ԃ�T�����B
�@�@�@�@UHF GEO-OK SADDLE
�@�@�@�@و�������
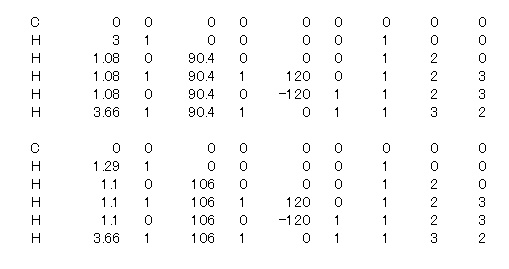
�@�@4-5�@��ܰ�ށ@�uTS�v�@�̎g����
| 1) |
���Ѵ�ٷޖ@�œ����T�Z�l�������l�Ƃ��Ă�萳�m�ȑJ�ڏ�Ԃ�^������W/�����M�����߂� |
| 2) |
���̔����ɂ��āApm3�ł͑J�ڏ�ԂɌ��ƂȂ�\���͓����Ȃ������B
MNDO �ɂ���TS�̌v�Z���s�Ȃ����B |
�@�@SYMMETRY UHF GEO-OK TS
�@�@و�������
| C |
0.00 |
0 |
0.0 |
0 |
0.0 |
0 |
0 |
0 |
0 |
| H |
1.29 |
1 |
0.0 |
0 |
0,0 |
0 |
1 |
0 |
0 |
| H |
1.10 |
1 |
106.0 |
1 |
0,0 |
0 |
1 |
2 |
0 |
| H |
1.10 |
0 |
106.0 |
0 |
120.0 |
0 |
1 |
2 |
3 |
| H |
1.10 |
0 |
106.0 |
0 |
-120.0 |
0 |
1 |
2 |
3 |
| H |
2.14 |
1 |
106.0 |
0 |
0,0 |
0 |
1 |
3 |
2 |
3�@1�@4�@5
3�@2�@4�@5�@6
�@�@4-6�@��ܰ�ށ@�uFORCE�v�@�̎g����
�@�@�@TS�œ���ꂽ�J�ڏ�ԂŐU�������߂�B���̐U���������ΑJ�ڏ��
�@�@�@SYMMETRY UHF GEO-OK FORCE
�@�@�@و�������
| C |
0.00 |
0 |
0,0 |
0 |
0.0 |
0 |
0 |
0 |
0 |
| H |
1.27 |
1 |
0.0 |
0 |
0.0 |
0 |
1 |
0 |
0 |
| H |
1.10 |
1 |
105.8 |
1 |
0.0 |
0 |
1 |
2 |
0 |
| H |
1.10 |
0 |
105.8 |
0 |
120.0 |
0 |
1 |
2 |
3 |
| H |
1.10 |
0 |
105.8 |
0 |
-120.0 |
0 |
1 |
2 |
3 |
| H |
2.14 |
1 |
105.8 |
0 |
0.0 |
0 |
1 |
3 |
2 |
�@3�@1�@4�@5�@6
�@3�@2�@4�@5�@6
�@�@���̐U��Ӱ�ނ��P��(2402i cm-l)����ꂽ�@���@�J�ڏ�ԂƔ��f�ł���
ROOT NO. �@1 �@�@�@2 �@�@�@3 �@�@�@4 �@�@�@5
�@�@�@�@6 �@�@�@7�@�@�@ 8 �@�@�@�@9 �@�@10
�@�@�@�@�@�@-2402 �@481�@ 481�@ 1167�@ 1167�@
1413�@ 1431�@ 1431�@ 1611�@ 3326
| ROOT NO. |
11 |
12 |
13 |
14 |
15 |
16 |
17 |
18 |
|
3326 |
3358 |
0 |
0 |
0 |
197 |
80 |
80 |
5.�@Ghemical�Ƃl�n�o�`�b�̃f�[�^����
�@5-1.�@Ghemical��MOPAC
�@�@�@0)�@Debian��į�߂̍���ɂ���u���ع���݁v�Ÿد��A���ع���݂̕��ނ�ؽĂ��\���B
�@�@�@�@�@�߲�����u�Ȋw�v�ɒu���Ʋ�ݽİق���Ă����Ă�ؽĂ��\���B�ugemical�v��د��BGhemical�̉�ʂ��\���B
�@�@�@1)�@��ʏ�ŕ��q��g�ݗ���(Ghemical�}�j���A���Q��)��B�ȕւȍ\���œK�����s�Ȃ�(���q�͊w�@)
�@�@�@2)�@�E�N���b�N��ؽĂ��\���̂ŁuFile�v���߲������ƉE��ؽĂ��\���B
�@�@�@�@�@����ؽĂ���uExport�v��د��A̧�ُ�������MOPAC
internal���د��A������������͂��Ăn�j��د�����B
�@�@�@3)�@Ghemical�����(�l�n�o�`�b�l��Z-matrix��̧�قɕۑ����ꂽ)�B
�@�@�@4)�@�ި��į�߂̍���ɂ���u�ꏊ�v��د��A�ިڸ�̓��e��\��Window���J���B
�@�@�@�@�@���y�̐ݒ肾���ިڸ�u�z�[���v��̧��FOR005���ۑ�����Ă���B
�@�@�@�@�@�l�n�o�`�b�V���̓t�@�C���gFOR005�h��2)�œ���ꂽZ-matrix���R�s�[
�@�@�@5)�@ Z-matrix�̑O�ɷ�ܰ��/���Ă���́A���point�l(�ω��l)����͂���"FOR005�h��ۑ�
�@5-2. M0PAC7����
�@�@�@0�j ��į�߂̍���ɂ���u���ع���݁v��د��B
�@�@�@�@�@�߲�����u����v�ɒu����ؽĂ��\��āu�[���v��د��B�����������Ẳ�ʂ��\���B
�@�@�@1)�@ �u/usr/lib/mopac7/mopac7 >FOR006�v����͂��邱�Ƃłl�n�o�`�b�V���N���B
�@�@�@�@�@F0R005�ɓ��͂��ꂽ�������ɂ��ķ�ܰ�ނœ��͂����v�Z���J�n
�@�@�@�@�@(���y���l�n�o�`�b�V��install��������ł͂l�n�oAC7�N���R�}���h���ިڸ�آ/��s��/lib/�����������V/��ɂȂ�A̧��
�@�@�@�@�@�F0R005����ިڸ�آ�z�[����ɂ������B�@�ިڸ�آ�z�[�����հ�ް�ިڸ��root�ިڸ��)�B
�@�@�@2�j MOPAC7�ł͓��o�̓t�@�C���̃t�@�C���������̗l�Ɋ��肵�Ă���B
�@�@�@�@�@
�@�@�@3)�@��肪�Ȃ���v���O�����I���B�G���[���o����Ώ�����B
�@�@�@4)�@ F0R005���ۑ�����Ă����ިڸ�Ɠ����ިڸ�Ɍv�Z���ʂƂ���̧��FOR006/FOR009/FOR010/FOR011/FOR012�ɏo�͂����B
�@�@�@5)�@�l�n�o�`�b�V���I������
�@�@�@6)�@FOR006��̧�ٖ����v�Z���e�ɕύX���ĊW̧�ق��ۑ�����Ă����ިڸ�Ɉړ����ĕۑ�����B
�@�@�@�@�@(�l�n�o�`�b�ł͓���̧��/�o��̧�ق̖��O��FOR005�`FOR012�Ɍ��߂Ďg���Ă���)
�@�@�@�@�@(���͗p/�o�͗p���ع����(�������v���O����)������č�Ƃ��ȒP�ɂ��邱�Ƃ��ł��邪�����ł͊�{�I�Ȏg�������q�ׂ�)
�@5-3. MOPAC���@Ghemical
�@�@�@1)�@ Ghemical���N������B
�@�@�@2)�@����2.6)�ō쐬�����t�@�C�����C���|�[�g����(���د��̧�ف����߰ĸد��A�uMOPAC
internal�v�� �د�)
�@�@�@3)�@��������̉摜�߂�
9.�@�Q�l����/URL
�@MOPAC7���g���n�߂鎞�ɂ́@4�j�A101�j �����ɗ����܂����B�@103)
�͂�����Ƃ����e�N�j�b�N��m�邱�Ƃ��ł��܂����B
| 1) |
�u�v�Z���w����v���f���@�u�k�л���ę̀�� |
| 2) |
�u�ʎq���w����v�đ�原�Y���@���w���l |
| 3) |
�u���q�O���@�l�n�o�`�b����ޯ��v�@����P�v���@�C���� |
| 4) |
�u�v�Z���w�����v�x�������@�ۑP |
| 5) |
�uApproximate Molecular Orbital Theory�vPople
& Beveridge�@McGraw-Hill |
| 6) |
�uThe molecular Orbital Theory of Organic Chemistry�vM.J.S.
Dewar�@McGraw-Hill |
| �V�j |
�u�c���������� �O�����v�������u�@�ĉj�� |
| �W�j |
�u�k���������V�X�e���m���H�n����v�B�����T�@�Z�p�]�_�� |
| �X�j |
|
| 101) |
�@http://jn1inl.blog77.fc2.com/blog-entry-1074.html�@�@�uLinux��MOPAC�����v |
| 102) |
�@http://www.mlb.co.jp/linux/science/�@�@�uLinux�ʼnȊw�����悤�v |
| 103�j |
�@http://jn1inl.blog77.fc2.com/blog-entry-1086.html�@����������k���� |
�@�@�@�@�@�@�@�@�@�@�@�@�@�@�@�@�@�@�@�@�@�@�@�@�@�@�@�@�@�@�@�@�@�@�@�@�@�@�@�@�@�@�@�@�@�@�@�@�@�@�@�@�ȁ@��
�y�Q�l�z �G ghemical �� MOPAC7�̎g������ł��B�@�g�n�l�n�A�k�h�l�n������̂��{���̕��q�O���@�ł����A�Q�l�܂ŁB
�y��Đ��̂l�n�o�`�b�v�Z
���傫�����q�̌v�Z��
���d���̌v�Z�œ����邱�Ɓ^���E
AL�̒蒅���@�ŗL�x�N�g���v�Z���ʂ����čl����A�@�y��Â���/���|�����鎞�ɔ����ߕ��͂����ӂ�
�����̋����ׂ�
�g�b�v�y�[�W�ɖ߂�