− 原子数が少ない分子の場合は良く優れた機能があるが、Z-matrixのを部分的に変更する時は手間がかかかることを体感できる −
(下の説明に使った計算値は実際の計算値と異なる場合があります、適宜正しい計算をしてください)
1.WINMOSTARのインストール
①ダウンロード
URL:https://winmostar.com/jp/ (WINMOSTARのメイン日本語HP)から実行型ファイルをダウンロードします。
②インストール
項目1①で得られた実行型ファイルを実行します。インストールされ、デスクトップにWINMOSTARのアイコンが置かれ、コマンドプロンプトでMOPACが起動できる環境を自動的に作ってくれます(コマンドプロンプトでMOPACを起動するためにはコマンドプロンプトをMS-Windowsをデスクトップに置く環境を作るソフトとFORTRAN/C言語のコンパイラがあることが必然です。小輩のPCは以前に別件でこれをやっていたので、WINMOSTARが自動的にやってくれたのではないかもしれません。因みにコマンドプロンプトのWindowを開いてくれるソフトはcygwin、C言語のコンパイラはgcc、MOPAC6が書かれているFortran77のコンパイラはg77、Fortranには90と95もあり、MOPAC7はFortran90/95に対応しているようです。cygwin/C言語コンパイラ/Fortranコンパイラについては各々のインストールについてはInternetで検索してやってください。)。
③無償品の期間と計算できる原子数
無償品の使用できる期間は13ヵ月、分子の原子数は30個以下です(この点、リナックスのグラフィックソフトは少なくとも無償MOPACの最大原子数でもできます)。
④WINMOSTAR使用方法
日本語のマニュアルがあると思います(小輩は読んだことありませんが)。以下に焼成反応に関するMOSTARの使い方だけを紹介します。
2.分子を描く
①平面で分子骨格原子を描き結合を描く
・項目1②で作られたWINMOSTARのアイコンをダブルクリックすると、WINNMOSTARが起動する。
・分子描写Windowには緑色の炭素原子と黄色の水素原子が単結合した化合物が描かれている。
・炭素原子の方を酸素原子に変えるため、このWindowの右上の「H 1∨」と表示されているボックスをクリックすると諸元素がロールするので酸素を選んでクリックする。ボックスは「O 8∨」の表示になる。このボックスの右に「Chng」と表示されているボックスをクリックする。原子は赤い酸素になる。
・編集をクリックするとコマンドのリストが表示される。この下の方にZ-matrixのコマンドがある。ここにポインタを置くと更にリストが表示され、この中のダミー原子追加のコマンドがあり、これをクリックする。この操作によって原子の番号2に原子名Xのダミー原子が登録される。このダミー原子はMOPACの最適化計算時に原子達が一直線に並んで計算が停止することを防ぐため。
・表示されている炭素原子を酸素原子にした時と同様な操作で原子をAl(アルミニウム)にする。
・予め元素名を「O 8∨」にしておく。「編集(E)」をクリックし、リストから「原子」、「原子追加」をクリックする。<原子を置く位置をクリックしてください>と表示されるので原子Alの右上当たりをクリックする。「Chng」をクリックすると新しく作られた原子が酸素(O)になる。アルミニウム原子をクリックすると、この原子が太線の円で囲まれ、新しく作った酸素原子が細線の円で囲まれる。分子描写Windowの上にある深緑色の○−○のアイコンをクリックすると結合が描かれる。
・同様たやり方で各原子を描き続けて、次のような平面化学式を描く。
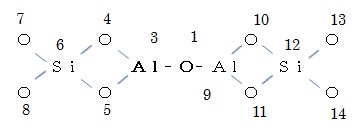
②水素原子を描く
「編集(E)」をクリックするとコマンドリストが表れる。「水素付加」「全原子」をクリックすると付加可能な水素が描写される。
3.最適構造の探索 (平面の分子化学式 → 3D分子構造)
①Z-マトリックスを完成させる
・WINMOSTAR画面の右に白地の部分が4つに区画されている。その真ん中の区画がZ-matrix。Z-matrizの下の深緑区画がZ-matrixのフラッグを修正するための区画。この段階では原子1〜3を除き、全て1であることを確認する。
②キーワードを書く
白地の一番上の区画がキーワードの区画。1行目に、「am1 」と入力する。 (遷移状態計算と合わせるため、am1を使用)
③MOPACの実行
・「計算1(C)」をクリック。コマンドリストが表れる。
・「(2)MOP7W70実行」をクリック。3D分子構造が最適化される。
4.遷移状態の探索
①水分子の入力
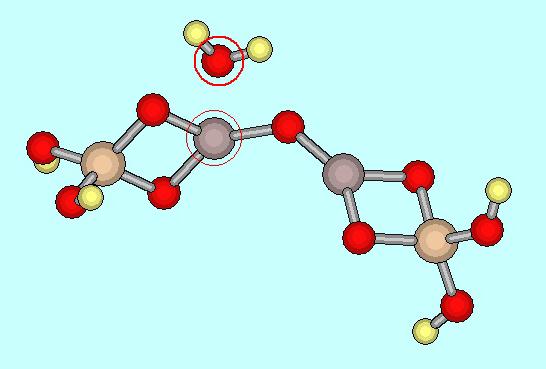
分子描写Window上でドラッグすると分子が自由に回転する。
原子1と原子3が反応しやすい場所に水分子を描く(項目2①と左図参照)。
②キーワードの入力
1行目に「am1 geo-ok」と入力します (遷移状態探索で収斂性がいいam1を使用)
− 加水分解の反応は水の2原子が協奏的にカオリナイト脱水化合物中のアルミニウム原子と酸素原子に反応するものなので、ディールスアルダー反応と同様にキーワードに「symmetry」を加えて試算したが収斂に至らなかった −
③Z-matrixの入力
水分子と反応に寄与する酸素原子とアルミニウム原子のフラッグを「1」として、他の原子のフラッグを「0」とします。但し、水分子の酸素原子の原子間距離のフラッグを「-1」とします。具体的には次の様になります(Z-matrixの作成方法がガチガチで面倒くさいのがWINMOSTARの欠点です)。
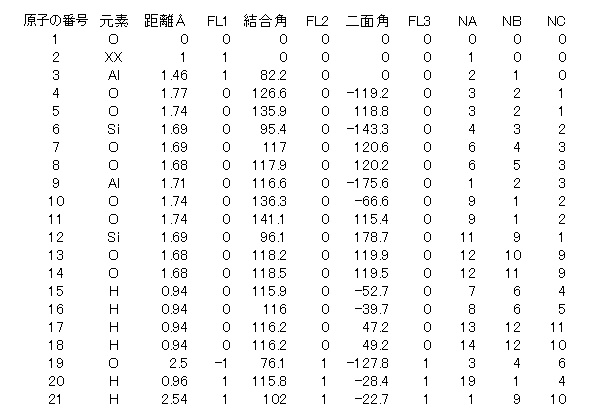
(WINMOSTARでは上表はきれいに表示されます。一般にMOPACでのZ-matrix、キーワードは空白字(複数空白でも可)で値が区切られ、上表のように列が蛇行しても計算してくれます)
④ミニマムエネルギーの計算のための入力
白地の最下段の区分に「2.5 2.4 2.3 2.2 2.1 2.0 1.9 1.8 1.7」と入力します。
⑤MOPACの実行
・「計算1(C)」をクリック。コマンドリストが表れる。「(2)MOP7W70実行」をクリック。
・計算開始に結果を保存するファイル名を聞いてくるので入力する。
・これで0.1Å刻みの最適分子構造と生成エネルギーが計算される。
⑥MOPACの計算結果
0.1Å刻みの最適分子構造で計算した生成エネルギー結果を次に示す。
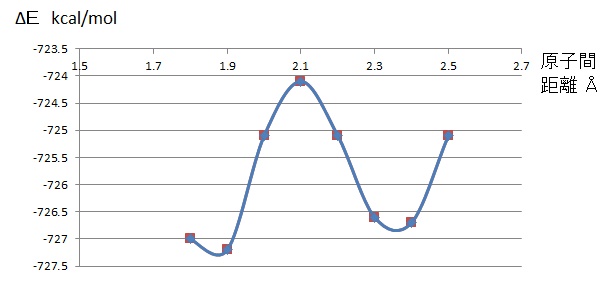
2.1Å付近に生成エネルギーのピーク(極大値)が見られた。
(原子間距離が1.8Å以下になると生成エネルギーが急激に高くなる。これは水分子の構造が異様に変形するため。上記項目2①の化学式以外の化合物の場合、急激な生成エネルギーの急激な増加の勾配の中にピークが肩として表れる。そのため正確な遷移エネルギーを得ることができないと考えられる(紫外線分析のチャート解析と同様な手法を使えばできるかもしれないが))
5.遷移エネルギーの計算
①計算1(C)」をクリック。コマンドリストが表れる。「インポート」「Animation(arc)」をクリック。
②ファイル名を聞いてくるので項目4⑤で入力したファイル名を選ぶ。0.1Å刻みの生成エネルギーとグラフが表示される。表示されたグラフの最大値(極大値)をクリックする(この機能がWINMOSTARの長所です)。
③キーワードに「am1 TS geo-onを入力する。
④水の酸素原子の原子間距離フラッグを「1」にしてMOPACを実行
⑤計算結果から生成エネルギーを読取る
⑥項目5⑤の生成エネルギーと加水分解物の生成エネルギー×2の差を計算すると遷移エネルギーが得られる。
6.計算結果が遷移状態のものであることの確認
①キーワードとして「am1 geo-ok UHF force」を入力する。
②Z-matrixのすべてのフラッグを「1」にする(1〜3番の原子については座標があるところだけ)。
③ミニマムエネルギーの計算のために白地の最下段の区分に入力した値を消して空白にする。
④MOPACを実行する。
⑤結合振動数が計算される。今回の反応ではマイナスの振動数の結合は2個 (通常の反応の場合は1個)であることを確認する。
7.比反応速度の計算
①アーレニウスの式
反応定数kは次の式で得られる

ここで、A:頻度係数、 E:活性化エネルギー、 R:ボルツマン定数、 T:絶対温度
②比反応速度を求める式
半経験分子軌道法では、生成熱から種々パラメータを出しているため、精製エネルギーは比較的正確である。頻度係数は分子の振動数から求めることができ、ab initioで計算された例は多いが、半経験分子軌道法による計算結果は正確とは言えない。そこで温度T1とT2におけるk1とk2の比を次の計算式で求めた。
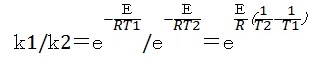
③予想反応時間の比較: 800℃の反応速度比を1、反応時間を30分とした場合、上記式を600、700、800、900℃で比較すると次の様になる
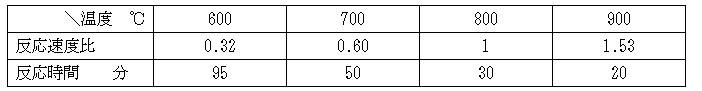
上記計算化学はあくまで試算で分析/測定機器に基づいたものでではないが焼成が700℃以下になると時間単位で焼成時間を要することがわかる。一般に550℃以下では反応しないと言われていることを考えると実際の活性化エネルギーは上記値より大きいことが考えられ、700℃以下では上表以上に焼成時間が長いと考えられる。
註) 上記ではカルサイトのモデル分子として、
の化学式(末端の酸素原子に水素がついている)の化合物を計算しましたが、同じ組成で原子3がSi(珪素)になり原子6がAl(アルミニウム)になったものや原子3と原子9両方ともSiになったものもある。これらの化学式の分子について上記と同様な計算をいろいろ試みたが、収斂しないか曲線の肩として表れてきれいな遷移状態のピークとならなりませんでした。従って、ここでは上記計算結果だけを示しました。以上
トップページに戻る